 glioblastoma, bladder cancer, and the bone cancer Ewing sarcoma.[2] Images courtesy of David Solomon.")
STAG2 is one of the most commonly mutated genes in (left to right) glioblastoma, bladder cancer, and the bone cancer Ewing sarcoma.[2] Images courtesy of David Solomon.
UCSF researchers have discovered that STAG2 – a gene commonly mutated in several human cancers – plays an essential role in DNA replication, revealing potential mechanisms for therapeutically targeting glioblastoma and other cancers.
As part of a multi-protein complex called cohesin, the STAG2 protein has been implicated in numerous cellular functions. The cohesin complex, which forms a ring-like structure that encircles DNA molecules, was first identified for its role in regulating the separation of DNA chromatids during mitosis (the process of cell division).
Published today in Nature Communications,1 UCSF neuropathologist and cancer biologist David Solomon, MD, PhD and colleagues identify a new role for STAG2. “We found that STAG2 is required for replication fork progression, which can be exploited as a therapeutic vulnerability in STAG2-mutant cancers,” said Solomon.
The replication fork is a structure that forms when DNA molecules are being replicated. Each strand of the DNA double helix is unwound and becomes a template for a new, matching partner strand, which is built and extended as the replication fork progresses.
The Solomon Lab reports that loss of STAG2, in normal cells, causes the replication fork to stall by disrupting interactions between the cohesin complex and DNA replication proteins. This leads to replication fork collapse and causes double-strand breaks in the DNA; consequent activation of DNA damage signaling pathways ultimately halts the cell cycle, thus preventing cell proliferation.
This leads to replication fork collapse and causes double-strand breaks in the DNA; consequent activation of DNA damage signaling pathways ultimately halts the cell cycle, thus preventing cell proliferation.
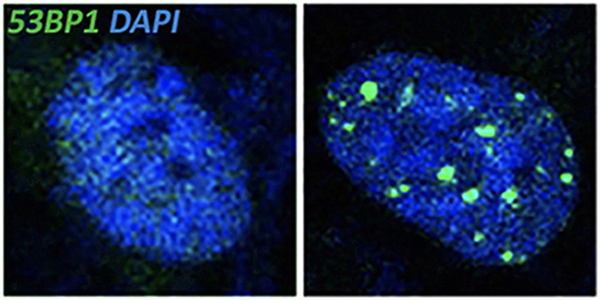
However, STAG2 inactivation behaves differently in tumor cells. “STAG2 inactivation is often associated with TP53 mutations, and in some cancers such as Ewing sarcoma, the combination of these mutations is associated with worse prognosis,” said Solomon.